La retinite pigmentosa: cause, sintomi ed ereditarietà

La retinite pigmentosa (RP) è una rara malattia ereditaria che comporta la lenta e progressiva degenerazione della retina dell’occhio, sensibile alla luce. La RP potrebbe anche causare cecità.
Se si sospetta la presenza di retinite pigmentosa, è probabile che si eseguirà un test del campo visivo durante o dopo i controlli visivi di routine per determinare la portata della perdita della visione periferica.
Potrebbero essere necessari altri esami specialistici per determinare se è accompagnata dalla perdita della visione notturna o dei colori.
Sintomi della retinite pigmentosa
Solitamente i primi segnali della retinite pigmentosa si manifestano nell’infanzia, quando entrambi gli occhi ne sono tendenzialmente interessati. La visione notturna potrebbe essere scarsa e anche il campo visivo potrebbe iniziare a restringersi.
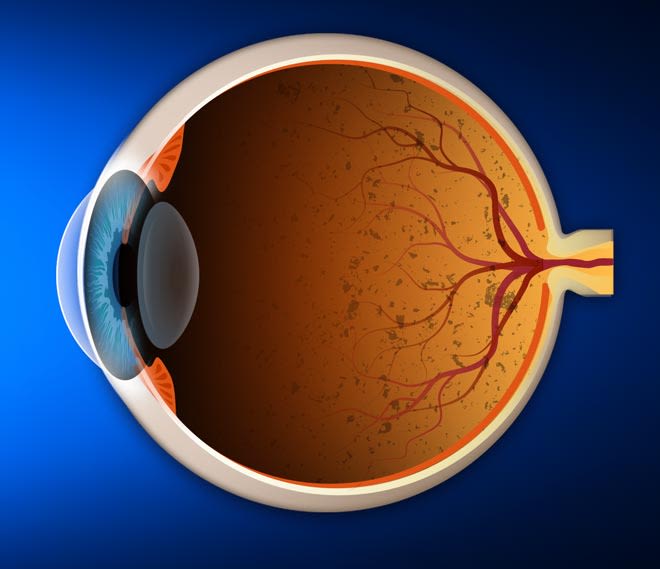
L’insorgenza della RP è accompagnata da un graduale deterioramento delle cellule fotosensibili responsabili della visione in presenza di poca luce (bastoncelli) e da una maggiore difficoltà a vedere di notte.
Nelle fasi avanzate della retinite pigmentosa, solo una piccola area della visione centrale resta attiva, insieme a una debole visione periferica.
È molto difficile prevedere la portata della perdita della vista o la velocità con cui progredirà la retinite pigmentosa. Il vostro oculista controllerà la salute delle cellule retiniche ed eseguirà dei test per determinare la vostra capacità visiva.
A un certo punto potrebbe consigliarvi di guidare solo di giorno o lungo strade ben illuminate di notte. Potreste infine non riuscire più a vedere così bene da poter guidare.
Quali sono le cause della retinite pigmentosa?
La retinite pigmentosa non è considerata una singola patologia ma come un gruppo di disturbi che interessano le cellule fotosensibili della retina che sono alla base della visione. Le cause della retinite pigmentosa non sono molto note; si sa solo che è una patologia ereditaria.
Questa patologia oculare è associata ad almeno 32 geni diversi che controllano tratti distintivi ereditati in molti modi diversi. A volte il tratto genetico è dominante ed è probabile che sarà trasmesso a un figlio nel caso in cui un genitore soffra di RP. Altre volte il gene della retinite pigmentosa è recessivo e potrebbe non manifestarsi per diverse generazioni prima di colpire un membro della famiglia.
Ciò significa che anche se vostra madre o vostro padre non soffre di retinite pigmentosa, potreste ereditare questa patologia oculare se almeno un genitore ha un gene alterato associato a questo tratto. Di fatto, circa l’1 percento della popolazione è considerato portatore di tendenze genetiche legate alla retinite pigmentosa.
Negli Stati Uniti, la retinite pigmentosa colpisce una persona su 4.000. Se è dominante, questo gene si manifesta con maggiore probabilità quando il soggetto ha circa 40 anni. Se è recessivo invece tende a manifestarsi per la prima volta all’età di 20 anni.
Cure per la RP
Al momento non esiste cura per la retinite pigmentosa.
Numerose aziende stanno sviluppando delle protesi retiniche (a volte chiamate occhi bionici) e altre cure innovative che si stanno rivelando promettenti nell’offrire o conservare una parte della visione utilizzabile nelle persone colpite da RP.
Argus II Retinal Prosthesis System. Second Sight Medical Products ha sviluppato l’Argus II Retinal Prosthesis System per pazienti resi ciechi dalla retinite pigmentosa e altre patologie degenerative della retina.
L’Argus II System include una minuscola videocamera integrata in uno speciale paio di occhiali. La videocamera è connessa a un piccolo dispositivo wireless indossato dal paziente che trasforma le immagini video in segnali elettrici trasmessi poi via wireless alla protesi all’interno dell’occhio.
L’impianto utilizza queste informazioni per stimolare le rimanenti cellule sane presenti nella retina e le informazioni visive vengono trasmesse dal nervo ottico al cervello, dove vengono recepite come stimoli luminosi. Il paziente impara a interpretare questi stimoli in modo da poter distinguere il profilo degli oggetti.
Secondo uno studio clinico condotto su 30 soggetti resi ciechi dalla RP o da altre patologie alla retina sottoposti a trapianto del dispositivo Argus II in 10 centri di chirurgia oculare in tutto il mondo, tutti i pazienti hanno ricevuto da Argus II degli stimoli visivi e molti hanno mostrato miglioramenti significativi nelle capacità motorie, quali la capacità di seguire delle linee, aprire porte e finestre ed evitare oggetti.
Due soggetti a cui era stato impiantato Argus II sono stati in grado di leggere brevi frasi, superando le aspettative dei ricercatori e i progressi visivi ottenuti da numerosi pazienti sono rimasti invariati durante un periodo di controllo di almeno due anni.
Retina Implant AG è un’altra azienda di dispositivi medicali che sta sviluppando un impianto retinico per chi soffre di grave perdita della vista a causa della retinite pigmentosa.
La tecnologia dell’impianto Alpha IMS dell’azienda tedesca è composta da un microchip quadrato in silicio di 3 millimetri quadrati che viene impiantato chirurgicamente sotto la retina, vicino alla macula. L’impianto contiene 1.500 cellule pixel, ognuna contenente un fotodiodo, un amplificatore e un elettrodo che trasmette i segnali elettrici al nervo ottico.
Le cellule pixel sostituiscono i naturali fotorecettori della retina, danneggiati dalla RP. Quando la luce colpisce i fotodiodi della protesi, crea un impulso elettrico che viene inviato al nervo ottico e all’area del cervello deputata alla visione.
Secondo l’azienda, poiché la protesi fotosensibile non può riprodurre il complesso processo di percezione visiva avviato dai fotorecettori naturali della retina, i pazienti dotati di questa protesi non possono visualizzare gli oggetti in modo nitido, ma sono in grado di localizzare le fonti luminose e gli oggetti fisici.
I risultati degli studi clinici sull’impianto Alpha IMS della Retina Implant AG hanno mostrato che i pazienti ciechi con RP sottoposti a impianto erano in grado di riconoscere i volti e leggere i cartelli sulle porte. Altri pazienti riuscivano a riconoscere gli oggetti e le espressioni facciali, nonché a leggere parole e vedere i punti su una coppia di dadi.
Terapia di stimolazione elettrica. Secondo i rappresentanti di Okuvision, un’azienda produttrice di dispositivi medicali fondata da Retina Implant AG, nei pazienti con RP in fase iniziale e intermedia la terapia di stimolazione elettrica (electrical stimulation therapy, EST) dell’occhio potrebbe aiutare a conservare la visione che andrebbe altrimenti persa a causa della malattia.
In uno studio clinico condotto su 24 pazienti con retinite pigmentosa in stadio iniziale o intermedio, i soggetti che avevano ricevuto una piccola quantità di corrente elettrica sulla retina tramite un minuscolo elettrodo mostravano un miglioramento significativo del campo visivo rispetto a quelli non sottoposti a stimolazione.
Secondo i ricercatori lo studio suggerisce che una stimolazione elettrica controllata della retina consente il rilascio di fattori della crescita che potrebbero ritardare la degenerazione della retina causata dalla RP.
Altre cure. Tra le altre possibili cure per la RP ci sono l’impianto di capsule contenenti un farmaco a rilascio controllato che potrebbe aiutare a conservare o prolungare il funzionamento della retina, terapie a base di vitamina A e altri antiossidanti per ridurre il danno alla retina e terapie geniche mirate a trapiantare i geni normali nelle cellule retiniche così da sostituire quelli mancanti o difettosi e bloccare o invertire il decorso della retinite pigmentosa.
Terapia adattiva e ausili per ipovisione
L’intervento repentino con una terapia occupazionale potrebbe aiutare ad affrontare i cambiamenti nella vita quotidiana dovuti alla RP, perché è più facile adattarsi a un calo della vista nelle fasi iniziali.
Chi soffre di retinite pigmentosa potrebbe anche utilizzare ausili per l’ipovisione che ingrandiscono e illuminano gli oggetti a casa e negli ambienti di lavoro.
Pagina pubblicata in mercoledì 8 dicembre 2021






